Leaderboard




Popular Content
Showing content with the highest reputation on 03/10/2021 in all areas
-
^^^^ That is what many of us are doing as part of our field work. So far as the "missing" skeletons, can you imagine the questions that could be answered by running DNA tests on the marrow, etc that might remain in the bones? Wow, treasure chest of answers.2 points
-
Maybe what we see aren't really eyes, but exhaust vents for a coal-powered furnace that operates a steam engine within the torso of the creature. Skepticism is welcome, but even good things can be taken a couple steps too far.2 points
-
Hi everyone - my name is Stephanie and I grew up in the PNW and currently live in Northern Alberta. I have had a couple encounters with Sasquatch type beings during my life. I am looking forward to discussing and continuing my journey towards increasing my knowledge of these beings.2 points
-
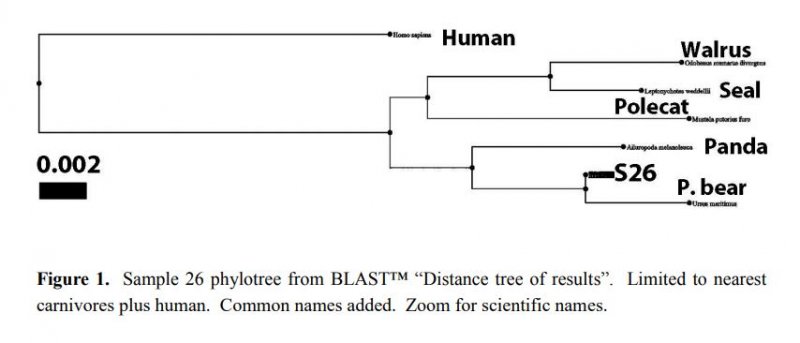
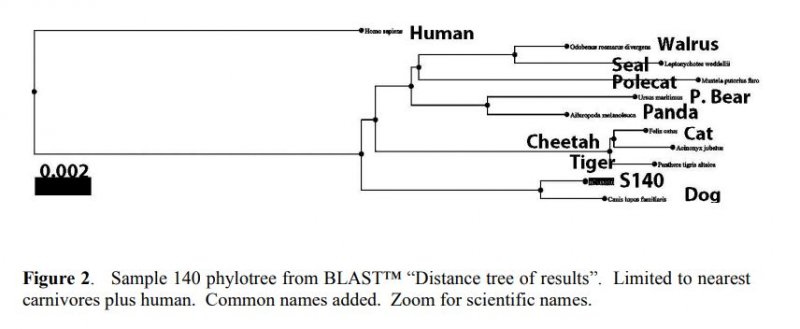
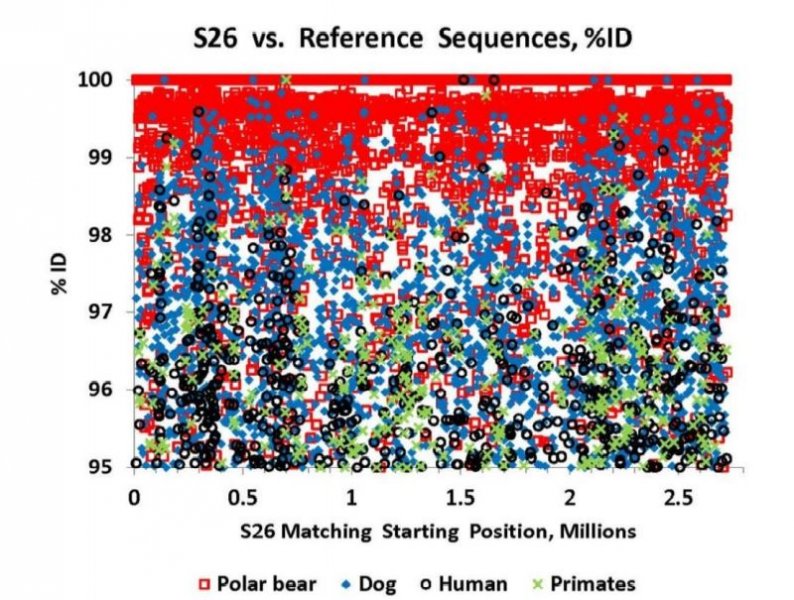
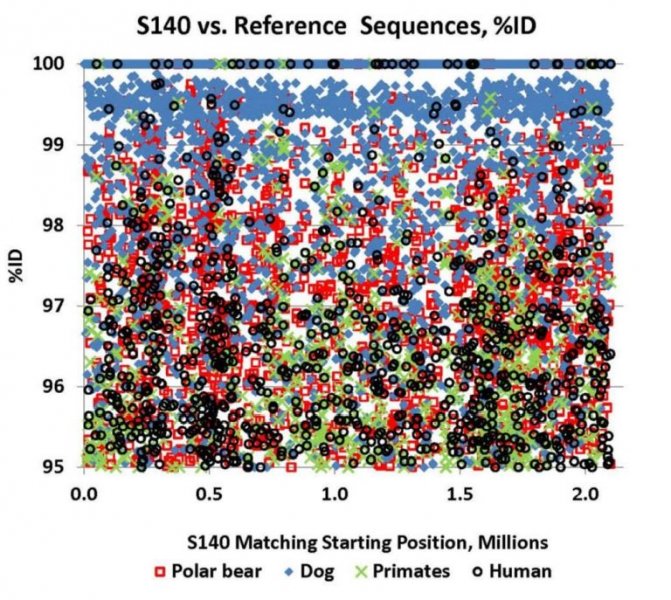
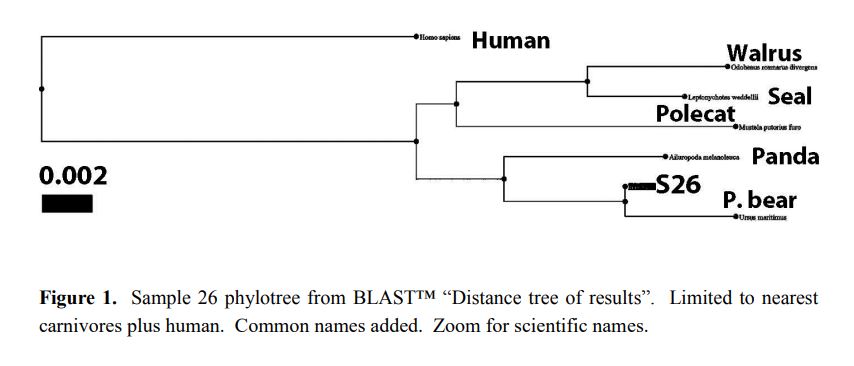
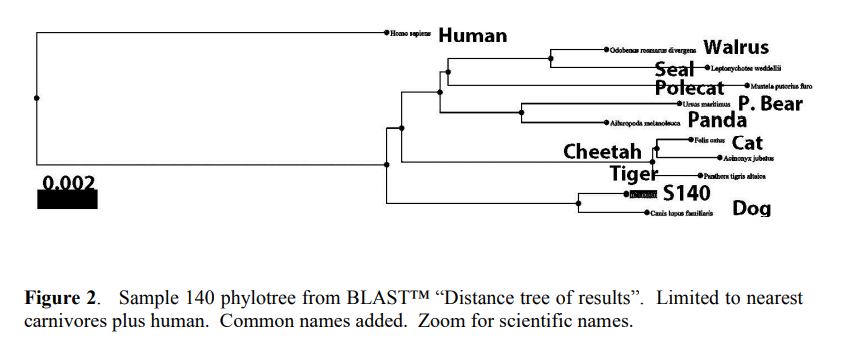
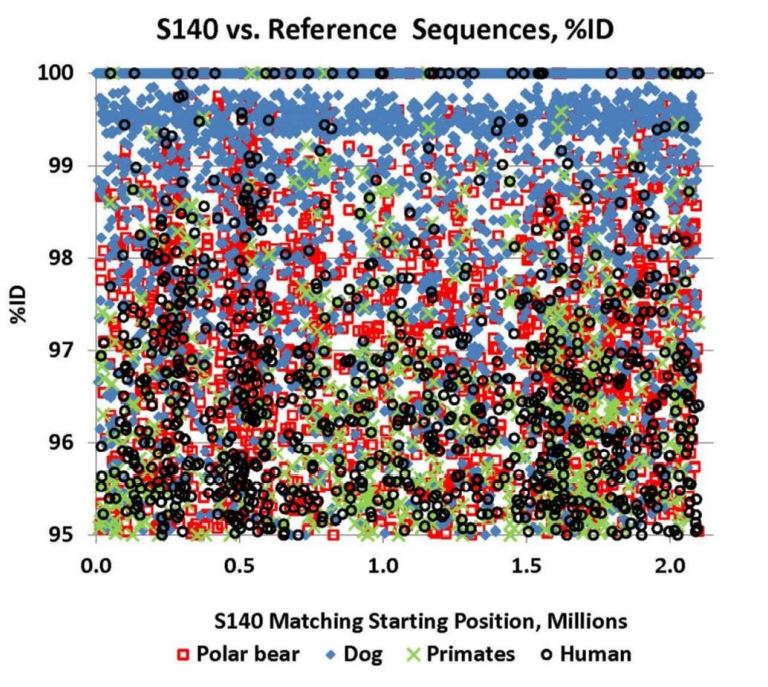
Reprinted with Permission 3/9/2021 - @hvhart sorry it took so long! The RELICT HOMINOID INQUIRY 5:8-31 (2016) Research Article DNA AS EVIDENCE FOR THE EXISTENCE OF RELICT HOMINOIDS Haskell V. Hart Canyon Lake, TX 78133 Correspondence to: Haskell V. Hart, email: hvhart@swbell.net © RHI KEY WORDS: sequencing, bigfoot, sasquatch, DNA, analysis ABSTRACT DNA sequencing methods available to the wildlife biologist or forensic anthropologist are briefly summarized. Their recent applications to potential relict hominoid samples are critically reviewed. Guidelines for sample collection and transport, analytical method selection, and interpretation of results are presented. None of the six published DNA studies to date have yielded any credible evidence for the existence of a relict hominoid. INTRODUCTION For information content, specificity, and universality of application, DNA methods of analysis are unbeatable among individual identification methods. With the advent of new (next generation) technology, whole nuclear genomes can be sequenced in weeks rather than years. Costs have also declined dramatically. The field of criminal forensics now holds DNA methods above all others in individual identification cases. For nearly fifty years following the famous 1967 Patterson-Gimlin Film (PGF) the only evidence for the existence of a relict hominoid (RH), which includes sasquatch, bigfoot, yeti, yeren, almasty, yowie, orang pendek, and other “large hairy ape-men” worldwide, was based on eyewitness accounts (numbering in the thousands), footprints, vocalizations, and a very few other videos and pictures, none as convincing as the PGF. Even the PGF itself is still debated as to its authenticity. Lacking the holy grail of a holotype specimen, the field was ripe for the application of new technologies. To date, several different DNA analysis (sequencing) methods have been employed on potential RH samples. The samples included hair, toenail, blood, tissue, and saliva, usually collected without documentation of the samples’ origins, i.e. no photograph, video, or even a personal observation that directly ties a sample to its origin (attested sightings in the area are no substitute). These analytical methods are summarized here. A more detailed description of each method can be found in Linacre and Tobe (2013). The very first application of DNA analysis to the problem of identifying a purported RH hair sample was presented as an April Fool’s joke in which the alleged “yeti” sample of interest turned out to be an odd-toed ungulate, most likely a horse, Equus caballus (Milinkovitch et al., 2004). Coltman and Davis (2005) reached a similar conclusion on a hair sample from the Yukon Territory; this time it was from an American bison (Bison bison). However, in February, 2013, after much advance publicity, Ketchum et al. (2013) self- published their ostensibly landmark paper claiming to have proven the existence of sasquatch based on 111 samples collected from 14 states and two Canadian provinces, which were subjected variously to both mitochondrial and nuclear DNA analyses as well as microscopy, both optical and electron. Their conclusions were that: “…the species possesses a novel mosaic pattern of nuclear DNA comprising novel sequences that are related to primates interspersed with sequences that are closely homologous to humans,” and “…that they are human hybrids originating from human females.” The only other extensive potential RH DNA study was by Sykes et al. (2014). Thirty hair samples, many of historical interest, were subjected to “rigorous decontamination” and sequenced in the 12S rRNA mtDNA gene. All samples matched known species of animals, including one of Ketchum’s (which turned out to be a black bear). Nothing that could be called a RH was reported, the closest being one modern human hair. After introducing relevant DNA analyses, their past applications to potential RH samples will be examined in detail. This review was prompted by the lack of any such thorough evaluation, in spite of the intense controversy surrounding some of the research. Only results and conclusions will be discussed; analytical protocol and laboratory practices are beyond the scope of this review, but are found in the original reports. MITOCHONDRIAL DNA Description of methods Inside the mitochondria of each cell are multiple copies of a circular double stranded DNA, which in humans has 16,568 nucleotide base pairs (bp). Since the sperm contributes no mitochondria, one’s mtDNA is inherited from the mother, and only a daughter can pass it on to grandchildren. Thus, it is a record of maternal inheritance. Humans do not all have identical mtDNA, though they are more than 99.5% identical. The differences, called mutations, are used in forensics and genealogy to establish maternal relationships. The human mitochondrial genome has 16 main genes and 22 short t-RNA genes (one for each amino acid). A few of the main genes are used to distinguish among different species. Although these important genes are conserved among mammals, they have accumulated distinct differences (mutations) between species. Mitochondrial methods of analysis have a copy number advantage over nuclear DNA methods; there are 1k – 10k mtDNA copies per cell, but only two nuclear DNA copies (one from each parent), except for the single X and Y chromosomes in males. When only a small amount of DNA is available (e.g. in a single hair), a mtDNA method is more likely to succeed because of this copy number advantage. Whole mtDNA Genome. The whole human mtDNA genome (all 16,568 base pairs) is sequenced when detailed comparisons are made between individuals within and between populations. It is the most time consuming of mtDNA methods, but can be done by a commercial laboratory, for humans, for about $200. If sufficient sample amount and funds are available this is the preferred method; it includes all the sequence regions below and more. All mitochondrial methods can be applied to nonhuman samples as well, with the use of universal or species specific primers. HVR-1 Region. In humans the Hypervariable Region 1 occupies positions 16024-16383 on the human mtDNA genome. Often a somewhat shorter segment is sequenced. As the name implies, this noncoding, control region has relatively more mutations per nucleotide than the coding region of mtDNA. It can therefore be used to get a super haplogroup or clade for an individual, but not a complete haplogroup, which may be enough to distinguish them from another individual. It has pitfalls, however, especially when a mixture (contamination) is involved, as we shall see later. Commercial laboratories will sequence your HVR-1 for about $100 (sometimes including HVR-2), which may be enough information to satisfy a genealogist. It was used by Ketchum et al. (2013) as discussed below. HVR-2 Region. In humans the HVR-2 Region occupies positions 57-372 in the mtDNA genome. It is somewhat less descriptive than HVR-1, but can be a confirmation of a haplogroup/clade. It was only rarely used by Ketchum et al. (2013). Cytochrome b. The Cytochrome b (cyt b) gene occupies positions 14747-15887 in the human mtDNA genome. This conserved gene is widely used in wildlife forensics to identify a species, genus, or family, but not individuals within a species (or genus or family). It was used by Ketchum et al. (2013) and independent laboratories as discussed below. 12S rRNA. The 12S rRNA gene occupies positions 648-1601 on the human mtDNA genome. This conserved gene is also useful in identifying a species, genus, or family. It was used by Sykes et al. (2014) as discussed below. Cytochrome c oxidase I. Cytochrome c oxidase I occupies positions 5904-7445 in the human mtDNA genome. Although it has not been used in RH investigations to date, this conserved gene also distinguishes among species (usually), genera, or families, in a manner similar to cyt b and 12S rRNA. A key factor in employing these methods is the selection of primers, which determine the segment of the gene to be sequenced. Primers are short DNA sequences which must match (as a complement) a specific short segment of the target sequence at its beginning and end, respectively. Primers can be universal for all mammals, or they can be specific for a species, genus, family, or order. Milinkovitch et al. (2004) A sample of hair obtained on the Matthiessen Expedition of 1992 to the Himalayas (Matthiessen and Laid, 1995) was the first purportedly RH sample to be analyzed by DNA methods. Using the conserved (universal) primers L1091 and H1478 of Kocher et al. (1989) the authors amplified by polymerization chain reaction, PCR, and sequenced a 417 bp segment of the mitochondrial gene 12S rRNA. They found that this purportedly Mehti (yeti) sequence matched a horse (Equus caballus), and they produced a convincing phylotree of closely related - zebra (Equus grevyi), kulan (Equus hemionus), donkey (Equus asinus), related - three species of rhinoceros, and unrelated - chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), human (Homo sapiens) - animals. Primates were very distantly related to the sample and the Equus. This was a very good beginning as to experimental methodology and interpretation of results, and not to be foreshadowed by the April Fool’s title and the conclusion that “extensive morphological convergences have occurred between yeti and primates,” which was not proven. Coltman and Davis (2005) Strands of hair were found near where a “large bipedal animal” was sighted. These were extracted, and a 429 bp fragment of HVR-1 was amplified and sequenced with conserved mammalian primers. The phylotree resulting from a BLAST™ (Altschul et al., 1990; Madden, 2003) search showed identity with the American bison (Bison bison), with nearest relatives, wisent, water buffalo, cow, and yak. The authors reported that DNA extraction was particularly difficult due to extensive degradation, consistent with over-winter weathering and exposure to direct sunlight or the consequence of tanning, and definitely not consistent with recent separation from the source (University of Alberta, 2005). Ketchum et al. (2013) Whole mitochondrial genomes were sequenced for 18 samples, and only HVR-1 for 11 samples in Ketchum et al. (2013, Supplemental Data 2), which lists all mutations from rCRS (revised Cambridge Reference Sequence). Supplemental Data 2 has no footnotes or text explanations about a reference sequence or which samples were HVR-1 only, which had to be deduced after the fact. All results are summarized in Table 1 here. Table 2 based on Hart (2016b, Table 1) shows extra mutations (not indicated by haplogroup) from Ketchum et al. (2013, Supplemental Data 2) whole genome results, including two listings (ES-1, ES-2) that were off to the bottom right and were not given sample numbers and were not mentioned in their text. Thirty-five H1a samples from GenBank (with 2.37 extra mutations on average) were used to calibrate a Poisson Distribution (Di Rienzo and Wilson, 1991) of extra mutations. Any more than six extra (“private”, not implied by the haplogroup) mutations have a less than 1% probability of occurring in this population (Hart, 2016b, Table 1). Further examination of the extra mutations in Table 2 here shows that in S2, S26, S36, S39b, S44 and S46, most of these extra mutations could be attributed to a second haplogroup, i.e. a contamination. However, Ketchum et al., in their paper and publicly, steadfastly deny any contamination in any of their samples. According to their Supplemental Materials and Methods S1, they used water and ethanol to vortex hair samples, but any with attached follicle, or any blood (S140), tissue (S26, S28), saliva (S31, S36, S37), nail (S35), or tree bark (S81) samples could not be so treated without dissolving the target DNA as well. These include some of the most important samples of the study; three have chromosome 11 nDNA results (see below). Interestingly, some of these extra mutations (Table 2) are in common between samples and are rare, especially in combination, in humans but common among other primates (Hart, 2016c). Whether these samples have a RH origin is uncertain, but further sample collection and analysis in these geographical areas are merited. Further, of the 11 HVR-1 only samples: four samples (41, 42, 43, and 140) have one extra mutation each. Four samples (71, 81, 117, and 118) have two equally likely haplogroups with one extra mutation for each alternative. Another sample is simply misgrouped (33), and only two (95, 168) are correctly and uniquely haplogrouped, all according to Behar et al. (2007, Supplemental Table S2). Thus, only three of the 11 samples could definitely be considered as phylogenetically modern human by this very limited HVR-1 criterion. The other eight could be something slightly different or could be contaminated by a second haplogroup (Hart, 2016b). Using universal primers on S26, Khan and White (2012) found that a cyt b sequence matched black bear. Similarly, black bear control region primers yielded a black bear control region sequence. Human control region primers yielded a human sequence, 402 bp long which matched exactly GenBank accession JQ705199, which was determined to be haplogroup T2b3e by Hart (2016b) from the full mtDNA sequence. Kahn and White cautiously avoided specifying a haplogroup, but it matches the T clade from their description of its geographic origin (Caucasus) and distribution (Middle East and Eastern Europe).[1] Three “Bristle swab in lysis buffer” control samples, which were likely from the sample submitter, Justin Smeja (but unspecified), matched the primary sample, S26, in HVR-1 (423 bp sequence). Cassidy (2013) found human cyt b, HVR-1, and HVR-2 in one sample cut from Ketchum et al. S26. Primers were not specified. A second sample (also S26), undoubtedly with unspecified universal or black bear primers, yielded a black bear cyt b sequence. The published sequences, when queried in BLAST™ as a check, matched human for HV-1 and HV2 (first sample) and black bear cyt b (second sample) as reported by Cassidy. Cassidy made a conservative haplogroup call of T2, although his HV1 region mutations were the same as Khan and White’s and indicative of T2b3e. Further examination of the extra mutations for S26 in Table 2 here revealed that 11 of 16 were T2b3e mutations, consistent with the findings of Khan and White (2012) and Cassidy (2013). Thus, three independent groups found a T2b3e contaminant in S26, although Ketchum et al. (2013) did not acknowledge it. Based on mitochondrial and nuclear sequences (see below), the contaminant DNA is very likely from the submitter, Justin Smeja. The decontamination procedure of Sykes et al. (2014) removed this contamination (see below). Sykes et al. (2014) In response to a public solicitation by the Museum of Zoology (Lausanne, Switzerland) and the University of Oxford, 57 “hair samples” were received from museums and private collections around the world. Of these, 30 were selected for DNA analysis. After “rigorous decontamination” (unfortunately, no details were given), the samples were all amplified and sequenced in 12S rRNA according to Melton and Holland (2007), producing 104 bp sequences, which were queried in GenBank with BLAST™. Unfortunately, the “rigorous decontamination” procedure is nowhere described. Elsewhere, a coauthor (Melton et al., 2005) used three ultrasonic water washes. Results were as follows: two brown bear (Ursus arctos), six American black bear (Ursus americanus) including the Ketchum et al. S26, two ancient “polar bear” (Ursus maritimus, but see below), four horse (Equus caballus), four cow (Bos taurus), four dog/wolf/coyote (Canis lupus/latrans/ domesticus), two raccoon (Procyon lotor), one white-tailed or mule deer (Odocoileus virginianus/hemionus), one American porcupine (Erethizon dorsatum), one serow (Capricornis sumatraensis), one sheep (Ovis aries, but see below), one Malaysian tapir (Tapirus indicus), one human, and no other primates. Independently run BLAST™ queries of all 30 sequences (which are in GenBank), confirmed the Sykes et al. findings with three minor differences. The sheep also matched a Himalayan tahr (Hemitragus jemlahicus); both are in the Caprinae family. The sequence for both “polar” bears had one mutation in common with brown bears and one with polar bears. In fact, there were brown bears with polar bear sequences in GenBank. A subspiecies of the Himalayan black bear, Ursus thibetanus japonicas, also matched brown bear over this limited range of 104 bp. This is evidence of hybridization and a complex bear phylogeny (too complex to be encapsulated in only 104 bp) due to the relatively recent divergence of polar and brown bears. Hybridization of brown and polar bears was previously known (Hailer et al., 2012; Cahill et al., 2013) and was acknowledged by Sykes et al. (2014). In commenting on the Sykes paper, Edwards and Barnett (2014) proposed that the two Sykes samples in question were from the Himalayan brown bear subspecies (Ursus arctos isabellinus). However, all agree that the two samples in question are from one of two (or possibly three) recently diverged, hybridizing, closely related species of Ursus bears and nothing even close to a primate. Overall, the Sykes et al. (2014) paper demonstrates the power of just 104 well selected bases in distinguishing species, or very occasionally only genus or family. In retrospect, he might have resolved ambiguities in some samples if a longer segment (e.g. as was done by Milinkovitch et al. and Coltman and Davis, above), or perhaps a second gene, were sequenced. However, the importance of laying to rest with scientific evidence the claims of RHs potentially present in his samples cannot be overestimated. It would have been a valuable addition to the paper to have included the origin of each sample, so that future investigators can either avoid duplication or appropriately compare results. Hopefully, Sykes et al. will make this information available upon request. As in the cases of Milinkovitch et al. and Coltman and Davis (above) an open-minded, universal (conserved) primer approach is the most appropriate approach in the early stages of a totally unknown species investigation by DNA sequencing. If greater species detail is still necessary more species-specific primers can be used later, sample amount permitting. Clearly, the Ketchum et al. study would have benefitted from this universal primer approach. Sequencing “whole” nDNA genomes of a black bear (S26) and a dog (S140) would have been avoided, and likely many other samples would have shown nonhuman matches by mtDNA sequencing with universal primers. It seems unlikely that all 111 of their study samples collected in the woods would turn out to have human mtDNA as reported, unless, of course, they were contaminated. NUCLEAR DNA Description of methods The nucleus of each human cell, regardless of its function, contains 22 pairs of autosomal chromosomes, one from each parent, and two sex chromosomes, one from each parent, for a total of 46. A female inherits an X chromosome from each parent and is called XX. A male inherits one X chromosome from the mother and one Y chromosome from the father, and is called XY. Thus the Y-chromosome is the indicator of paternal inheritance, just as mtDNA is for maternal inheritance. Sperm cells, which contain only one set of chromosomes, can be either X or Y, which determines the sex of the fertilized egg. The unfertilized egg has a single X chromosome from the mother. There are about 3.5 billion base pairs in the human nuclear DNA genome. There are over 24,000 human genes and much more spacer and unused (“junk”) DNA in the nuclear genome. Other animals have different numbers of chromosomes (e.g. chimpanzees have 48) and different numbers of genes in each. However, related organisms have some conserved nuclear genes in common, sometimes with relatively minor mutations, just as in the case of mtDNA. Whole Genome De Novo. The whole genome sequence is the holy grail of DNA, but it may not be affordable, possible, necessary, or practical in every case. The first human genome took 10 years to complete, but more modern techniques have reduced the time to weeks. The process can be reduced to (1) breaking the DNA into bite-sized pieces (<1000 bp), (2) amplifying these (making many copies), (3) sequencing these millions of segments, and (4) piecing the segments together by matching overlaps by de novo (computer), which assumes no species, or by using a reference sequence of a closely related species, when known. A whole genome has not been obtained from any purportedly RH sample to date, in spite of the claim in the Ketchum et al. (2013) title. Reference Sequence. If a sample is known to be closely related to some species (e.g. two Ursus bears), the nDNA sequence of the reference species can be used as a template (complement) to sequence the unknown, by noting and resolving the SNPs (single nucleotide polymorphisms – which are mutations) between the two. The method is widely used in studying genetic defects in humans, where relatively very few mutations in specific locations are involved, and the bulk of the patient DNA is very normal, permitting a normal human template. This method was also used by Ketchum et al. (2013), inappropriately in retrospect. The method is not suitable for totally unknown samples. Short Tandem Repeats at Microsatellite Loci. In and among genes are segments that are short tandem repeats (STRs) of 3-6 nucleotide bases, such as AGCAGCAGC AGC… The DNA replication process appears to “stutter” here, but importantly, it produces different numbers of repeats in different groups of individuals of the same species, and these different numbers are inherited, one from each parent, as alleles. Determination of alleles at multiple “microsatellite loci” can identify an individual either from its own reference sample or from those of its parents, usually with high probability (assuming enough loci). The method is used in criminal forensics and population genetics, and was used by Ketchum et al. (2013). Unfortunately, the method requires that you know what species you are dealing with and what the lagging and leading strand sequences are in order to pick the correct primers to sequence the intervening STRs (number of repeats). The method is not suitable for totally unknown samples. Specific Gene Sequencing. In a manner similar to mitochondrial methods, primers can be selected to target a specific portion of a nuclear gene, usually to detect SNPs related to a specific phenotype (gene expression). Ketchum et al. (2013) used this method with several genes, as discussed below. Again, the method requires detailed knowledge of the specific species’ gene sequence to select appropriate primers. The method is not suitable for totally unknown samples. Bead Array Analysis for SNPs. This technique is designed for identifying SNPs between different samples of the same species. A good example of its use is identifying SNPs involved in a particular disease. A particular method (application) would involve, identifying and synthesizing short DNA segments of interest and attaching them as probes to very small (3-6 μm) activated silica beads in wells on a slide or microchip. Incredibly, these different sequences are attached to the beads as probes in many copies each on up to millions of known and controlled bead locations, one per SNP on the chip. The sample (target) DNA is transcribed to its complement (cDNA) and fragmented into small segments, each of which is labelled with a fluorescent dye. The labelled sample is then hybridized with the probes on the beads and the excess washed off. The chip is then scanned with a laser of appropriate frequency to detect the locations of the hybridized probes by fluorescence, and hence which specific SNPs of the sample are present. Unhybridized probes show no fluorescence at their locations. If two different dyes are used, one for each SNP (nucleotide variation) both can be determined in a single scan with two lasers (one for each dye), providing both probes were present, either on the same bead for increased efficiency, or on two beads for each SNP. The single dye method requires either two beads for each SNP or multiple runs with different SNP probes. Determining allele ratios is semi-quantitative and requires standards and underlying assumptions. The method is not suitable for totally unknown samples, except as a very expensive and complex way of matching an unknown sample to a very specific known species, with no indication of the species if there is no match. This was the Ketchum et al. (2013) approach: attempting to match unknown samples to human. Electron Microscopy. Scanning electron microscopy (SEM) is used to examine overall morphology of DNA. The challenge is that placing a molecule that likes water in a high vacuum can cause artefactual changes in morphology. “Environmental” SEM (ESEM) can minimize this effect by covering the sample in a thin layer of water vapor, enough to preserve some morphology, but not enough to cause serious deflection of the electron beam and attendant defocusing. Use of a reference sequence The three nDNA sequences for samples 26, 31, and 140 (Ketchum et al., 2013, Supplemental Data 4, 5, and 6) were the most significant and testable data in the entire paper. Unfortunately, their method used human chromosome 11 as a reference for the sequencing, thereby both greatly reducing the length of the resulting consensus sequences and biasing them toward only highly conserved human genes. Consequently, the sequences contained only 2.7M, 0.53M, and 2.1M bp each, 0.4 - 2% of chromosome 11, and less than 0.1% of the entire genome in each case. The preferred method for a totally unknown species would be the de novo method, which does not assume a particular species. If it had been used, the sequences would be much longer and would contain more representative genes, both conserved and nonconserved. Furthermore, the match to database sequences would likely have been more discriminating and easier to interpret. Ketchum et al. concluded that all three sequences were from an unknown male primate/human female hybrid, and that they contained a mosaic of both human and other primate segments. Sample 26 is a black bear (Ursus americanus). From searches of Genbank with BLAST™, using the whole S26 nDNA sequence as query, it was found that S26 matched human and other primates only 94-95%, but matched polar bear (Ursus maritimus) about 98-99%. Black bear sequences in GenBank were sparse and relatively short, but matched S26 100%. Over five different database sets, three in GenBank and two from the literature (Cahill et al., 2013; Cronin et al., 2014), S26 consistently matched black bear or polar bear about 98-99% and human and other primates 94-95%. At first sight, this may appear to be a small difference, but considering that the S26 sequence was referenced to human chromosome 11 and that the difference was consistent over five different DNA datasets, the conclusion is sound that S26 is a black bear (Hart, 2016a). Further, a phylotree constructed from S26 consensus sequence hits in the refseq_genomic database in GenBank showed S26 in precisely the correct taxonomic position for a black bear in relation to the many other families and orders of mammals. Figure 1 here is an abbreviated version of that phylotree. In contrast, the corresponding Ketchum phylotree (their Supplemental Figure 4) shows homology with a variety of primates, including human. Their average distance to S26 was 0.03 - 0.04 (3% - 4% difference). This phylotree matches the S31 human sample better and may have been mislabeled. The distance to the polar bear (the nearest black bear relative in the refseq_genomic database) was only 0.006 in our phylotree, and we used the same tree-generating software in BLAST™. Ketchum et al. correctly concluded that S31 is human. Most database hits were 100%ID modern human (Hart, 2016a). However, their phylotree, (their Supplemental Figure 5) is bizarre. It showed equally distant relationships to human and mouse (Mus musculus) and slightly more distant relationships to a chicken (Gallus gallus), a carp (Cyprinus carpio), 25 species of other boney fish, and 12 species of sharks. Nothing else! Where are the primates and other mammals? Clearly something went wrong there. Sample 140 is a dog (Canis lupus familiaris) or less likely a wolf or coyote, not a sasquatch. Since there is a wealth of dog DNA in GenBank, no other source was queried. Hits averaged 99% ID match to dog compared to 94% for both human and other primates (Hart, 2016a). A phylotree in Figure 2, constructed as above, shows S140 in a position of 0.005 distance from a dog, and appropriately related to other carnivores and distant from human, agreeing with accepted taxonomy. In contrast, the Ketchum et al. (2013) phylotree (their Supplemental Figure 6) only contained human and mice, no other mammals. Again, it does not support their conclusions. No search hit results were given by Ketchum et al. (2013) in support of their phylotrees. They queried the nucleotide database, but we chose the Reference Genomic Sequences (refseq_genomic) database for our phylotrees, because, although it has fewer species, it is guaranteed to have the corresponding genes (a complete genome) for a matching species. The nucleotide database is incomplete for most species, and in fact, until well after Ketchum et al. published, there were no polar bear sequences in this database and very limited black bear sequences. Instead of accepting relatively poor hits, they should have searched other databases in GenBank. Short tandem repeats The Ketchum et al. (2013) Table 5 shows the results of STR analysis of 16 human microsatellite loci in 14 samples. One sample, S28, shows alleles at 15 loci, all others failed to sequence or were off ladder (unknown allele) at five or more loci; most failed at many more loci. Both of their control samples sequenced normally. Interestingly, S31, and S140, were not among these samples; hairs from S26 were included as S25. The Amelogenin gene appears as Amel X on the X-chromosome and as Amel Y on the Y-chromosome. It is a sex determining gene in that the absence of Amel Y indicates a female. Both sexes have the Amel X gene. However, among 33 samples in Ketchum et al. (2013) Table 3, 12 failed to sequence at either locus, and seven sequenced at Amel Y only (a genetic impossibility). Moreover, of 13 samples listed in both Tables 3 and 5, six had inconsistent results between the two. With primers for 16 human loci, Khan and White (2012) got no human alleles for S26, but when they used 15 black bear primers they found normal black bear alleles at 14 loci.[2] The fifteenth, Amel Y, produced no alleles, so the sample is female. Cassidy (2013) only employed human primers on S26, and found that at 13 of 16 loci the alleles matched the control sample from the submitter, Justin Smeja, exactly. Three loci produced no signals, probably because of the relatively small amount of human nDNA. With this many loci sequencing, the probability that Justin Smeja contaminated the sample is very, very high. Interestingly, for duplicate S26 analyses, their results matched Ketchum et al. (2013) for Amel X and Amel Y (both present), but did not match on any of the other nine loci, although in six cases one of two alleles was in common. It does not appear that the same human DNA (if any) was sequenced by Ketchum et al. Human microsatellite loci were the same for Ketchum et al. (2013) and Cassidy (2013) and differed by two from those in Khan and White (2012). Specific gene sequencing Loci on the genes Amelogenin, MC1R, MHY16, and TAP1 were sequenced by Ketchum et al. (2013). These results are found in Tables 4, 6, 7, and 7 of their paper, respectively. Also, longer gene sequences found in Supplemental Data 3 and on the Sasquatch Genome Project (SGP) website, http://sasquatchgenomeproject.org/, were aligned with BLAST™ and form the basis for the following. Amelogenin. As mentioned above, both sexes have the Amel X gene. Of 27 samples tested with human primers (Ketchum et al., Table 3), 15 failed to sequence Amel X, and two other samples had unknown sequences. Sample 26 sequences from the SGP website matched dog for Amel X (99.46%) and matched polar bear (100%) and giant panda (Ailuropoda melanoleuca) 95.27% for Amel Y Exon 2 (Recall the paucity of black bear data). Five of six sequences from Supplemental Data 3 were less definitive. Although the S35 Amel X sequence matched human 99.51%, S26 Amel X and S43 and S44 “Amel” sequences (two for each, unspecified whether X or Y) had no matches in any GenBank database. MC1R. Of 26 samples sequenced (Ketchum et al., 2013, Table 6), only three samples (39b, 85, and 121c) matched a reference sequence over all 10 loci, mutations at two of which are involved in red hair, a commonly observed sasquatch phenotype. Nine other samples had 1-3 mutations. The remaining 14 samples either failed to sequence at two loci (7 samples) or gave unknown sequences (7 samples). Both control samples sequenced normally. The large number of anomalies observed indicates that most samples were not modern human or that insufficient DNA was extracted. Ostensibly to correlate red hair with mutations at two loci, this study found that the black and white hair of S26 had both mutations for red hair. Others have seen that the phenotype of red hair is likely controlled by multiple genes and does not always follow simple Mendelian genetics based on dominant and recessive genes (Starr, 2011; McDonald, 2011). A MC1R sequence from sample 25/26 from the SGP website matched human 100% (GenBank accession AB598380.1). MYH16 (My16). Ketchum et al. (2013) report that all samples tested matched human sequences, however, the specific samples were not identified. One My16 sequence on the SGP website (S26) and two (S35 and S37) from Supplemental Data 3 of Ketchum et al. (2013) matched human 100%. TAP1. Supplemental Data 3 of Ketchum et al. (2013) gives TAP1 sequences for samples 10, 26, 33, 35, 39b, 43, and 44. S26, S35, and 39b matched human (100%), S10 matched dog (99.18%), part of S33 matched human mtDNA (100%) the rest was unknown, and S43 and S44 matched nothing in GenBank. The unknown portion of S33 matched the reverse strand of S44. While we agree with Ketchum et al. (2013) Table 7 that S33 and S44 partially align, we disagree that S10 and S43 align at all (or even with a reverse strand of one). PNLIP. A S26 sequence from SGP matched human 100%. HAR1. A S26 sequence from the SGP matched polar bear 98.08% and giant panda 92.86%. SNP analysis by bead array Twenty-four samples were subjected to whole nuclear genome SNP bead array analysis (2.5 M SNPs), however, only 12 results were reported, presumably the 12 highest performers. Results in Table 1, taken from Ketchum et al. (2013, Table 8) show that no sample was above the nominal human threshold of 95% SNP match, and most were much less. One human control matched 99.63%, a second slightly degraded human control matched 97.15%. The second control was nonsterile blood left at room temperature in a moist environment for four days, which in no way replicates the conditions in the field, which might include heat, sunlight (incl. UV), rain, microbial, fungal, or viral attack, not to mention casual contact with other species (e.g. insects), all for potentially lengthy periods of time (at least five weeks for S26, for example, and unknown periods for many samples). The implication that the relatively high match of the degraded human control proves that the other samples with much poorer matches are not degraded human is false. It is impossible to say whether low % SNP match is a caused by degradation of a human sample, or the natural result of a nonhuman sample subjected to human probes. As mentioned above, the method is inappropriate for totally unknown samples. If, however, this method is used, it should at least include control samples of some known nonhumans to calibrate how far from human the study samples might be if not degraded. A good short list would be a chimpanzee, a dog, a bear, and a horse, for example. The chimpanzee would represent the lower limit % match for any human-like hominoid. Other animals should show even lower % matches, differing from human by an amount increasing with their increased genetic divergence. Also, notably lacking were study samples 31 and 140, for which “whole” nuclear genomes were sequenced. Had they been included, a human like result (except for possible degradation) for S31, and a much lower result for the dog would be expected. Comparing the results for S26, S31, and S140, would have shown that these are either different species, or are representative of different degrees of degradation, or some combination of both. The agarose gel electrophoreses of S140 with ethidium bromide staining showed extensive streaking (Ketchum et al., 2013, Figure 10), a telltale sign of degradation. S26 and S31 were relatively much cleaner, however. This test does not measure contamination, however. Of course, contamination can affect SNP results, too, for example the highest % match for S26 (the black bear) is probably due to its extensive human contamination, as mentioned above. Electron microscopy Conventional SEM with platinum shadowing (not ESEM) of S26 nDNA showed sections of single-stranded DNA intermixed with normal double-stranded DNA. A degraded human blood sample, the same one mentioned above, showed no sections of single-stranded DNA (Ketchum et al., 2013). Their conclusion that degraded DNA cannot show single strands and therefore such a feature is evidence of a novel form of DNA is false. Single-stranded DNA is not novel (e.g., Desai and Shankar, 2003; Lehtinen et al., 2008) and can be the result of degradation (Ward et al., 1985). SUMMARY TABLE OF KETCHUM et al. RESULTS Table 1 is an Excel® compilation of all Ketchum et al. (2013) DNA results, including additional independent lab results on S26 (Khan and White, 2012; Cassidy, 2013; Sykes et al., 2014) and different interpretations of results by Hart (2016a, 2016b). Column headings are frozen for comparing methods across samples, but sample numbers can also be frozen for convenience in comparing samples across methods. A table as massive as Table 1 can be intimidating to assimilate and to draw conclusions from. Others are encouraged to draw their own conclusions and publish them. Samples 8, 48, 50, 51, 52, 53, 70, 75, 76, 77, 79, 80, 84, 86, 92, 93, 101, 102, 104, 105, 107, 108, 110, 111, 112, 116, 119a, 119b, 119c, 119d, 120, 121a, 121b, 126, 127, 128, 129, 131, 133, 134, 135, 136, 137, and 139 appeared in the 111 sample master Table 1 of Ketchum et al. (2013) but were not analyzed or reported elsewhere in the paper. Their Supplemental Materials and Methods states, “Hairs without tissue or root material did not yield DNA in this study,” although the text states, “All 111 screened samples revealed 100% human cytochrome b and hypervariable region 1 sequences with no heteroplasmic bases that would indicate contamination or a mixture.” It is not clear which of these conflicting statements is incorrect, but numerous heteroplasmic designations are found in their Supplemental Data 2. Moreover, whole mitochondrial genome sequences with the most extra mutations from the haplogroup were seen to be mixtures of haplogroups (Table 2 here). Samples 3a, 4, 5, 7, 13, 19, 30, 39b, 71, 72, 106a did not appear in Ketchum et al. Table 1 but were analyzed by them and appear in Table 1 here. CONCLUSIONS The studies of Milinkovitch et al. (2004) and Coltman and Davis (2005) are models for this kind of research. They used universal primers over a long sequence, they properly queried their sequence against Genbank (yielding good matches), and they produced believable phylotrees of resulting hits. Their conclusions are irrefutable. No sasquatch was found. Likewise, Sykes et al. did the same, except phylotrees were not produced from the short 104 bp sequences and would not likely have shed additional light on their 100% matches. The few slight ambiguities between polar and brown bears, the genus Canis, two deer, and two sheep, were not at all detrimental to the overall conclusion that no primate was found in 29 samples, and that a human was the 30th. The human sample was also sequenced in the HVR-1 region and matched 100% the revised Cambridge Reference Sequence, so it is modern human. Public criticism that Sykes et al. “cherry picked” the samples to disprove the existence of sasquatch is probably unjustified, based on his published sample solicitation and selection protocols, although there are claims that some submitted samples were neither acknowledged nor reported. Of 57 samples submitted, two were not hairs, and 37 were selected for analysis based on “provenance or historic interest.” Only 30 of these produced sufficient DNA for sequencing. The “much ado about nothing” over the “polar bear” samples is overblown given the purpose of the paper, although it prompted some good bear phylogenetic work by Edwards and Barnett (2014). All agreed that the two samples were from bears, not a RH. Finally, by far the most controversial and difficult to assess work was done by Ketchum et al. (2013) and elaborated on in their SGP website. Based on all available data in Table 1, it is seen that of the 78 samples with data reported by Ketchum et al. (2013), only Samples 2, 11, 12, 21, 31, 37, 87, 90, 95 and 117 can be considered definitely human, but of these, 11, 12, 21, 87, and 90 have very limited analyses. All these are highlighted in yellow. Additionally, samples 28 and 35 are very likely to be human.[3] All other samples have too many anomalies and inconsistencies to be called human or even near human. These samples were repeatedly called “novel” and/or “hominin” by Ketchum et al., rather than the more common and likely cases of misprimed (wrong animal), or self-primed, due to degradation or contamination. These phenomena are well known for “ancient” DNA, which practically speaking, is any DNA not taken directly from a known live animal in hand. Further, if a series of analyses do not all point indisputably to human, the distance from human is not easily discerned from the discrepancies, especially if they are relatively many. To determine whether nonhuman species would amplify and sequence with the human Amel X and Amel Y primers used by Ketchum et al. in their Table 4, human amplicons were determined from their primers in Supplementary Data 12 of Ketchum et al. (2013) and searched with BLAST™ for matches to other species. The primers were first aligned against human reference sequences (e.g. NW_001842425.2) of the appropriate chromosome. The extreme base positions at the far end of each primer (5’ on down-strand, 3’ on up-strand) then defined the amplicon, which in every case was on the correct chromosome and matched the length listed in Supplementary Data 12. The string of bases between these extreme positions was then searched against the Reference Genomic Sequences Database. As a check, the same results were obtained using Primer-BLAST™. Amel X produced identical hit results for chimpanzee (Pan troglodytes) and pygmy chimpanzee (Pan paniscus); these should amplify and sequence. Gorilla (Gorilla gorilla gorilla) and the northern white-cheeked gibbon (Nomascus leucogenys) are questionable with four total primer mutations. No other species matched. In any case, a human match for Amel X indicates a species more recent than any of the great apes. Similarly, only the chimpanzee (Pan troglodytes) aligned the primers at the proper locations and produced an amplicon of the correct length (Supplementary Data 12) on the correct gene for Amel Y exons 1, 2, 4/5, and 8. Hence, any male primate between chimpanzee and human on the Evolutionary Tree of Life would be amplified and sequenced at exons 1, 2, 4/5, and 8, and no other, more distant, male species of primate or nonprimate would be amplified and sequenced with these primers. Therefore, we are assured that failure to sequence or unknown sequence for Amel X or for a male Amel Y cannot be due to an unknown primate or human hybrid more recent than the chimpanzee; they must signal a more distant species, primate or nonprimate. Conversely, a “human” match to an amplicon from these four pairs of primers can only be a human or some human-like primate more recent than the chimpanzee, nothing else. Not even the gorilla, the pygmy chimpanzee, gibbons, or the orangutan (Pongo abelii) would align or sequence with these four primer pairs (too many mutations vs. the primers). Most likely, similar results would be obtained for other genes. Therefore, any “*” or “FTS” in Table 1 here are not likely due to a primate more recent (less distant) than a chimpanzee. These are other animals, not “novel” or “hominin.” The most studied sample by far is S26 (probably because it contained the most DNA), said to be a “hominin” by Ketchum et al. (2013) in their conclusion, which applied to S31 and S140 also: “Analysis of whole genome sequence and analysis of preliminary phylogeny trees from the Sasquatch indicated that the species possesses a novel mosaic pattern of nuclear DNA comprising novel sequences that are related to primates interspersed with sequences that are closely homologous to humans.” Figure 3 shows this to be false. All refseq_genomic database hits >200 bp and >95%ID are plotted, for the four species. Clearly, the polar bear (the closest bear relative to a black bear in the database) is the best match over all 2.7 M bp of the S26 sequence. Human and primates are the poorest matches, and have very few hits above 99%ID, whereas polar bear hits are concentrated above 99%ID. Three other independent laboratories showed S26 to be a black bear, the only extant bear in California, the collection location of S26. Human or human like results for other DNA analyses suggest contamination, especially by Justin Smeja, the sample collector, whose haplogroup – T2b or possibly T2b3e matched the S26 human contamination found experimentally by the other two forensic laboratories and deduced from the full mtDNA sequence in Table 2 here. Sample 31 is human by all accounts. It is also contaminated by fungus and bacteria. If, in fact, a sasquatch origin could be claimed it would require a nDNA sequence over more than just the reference chromosome 11 reported by Ketchum et al, which showed very few SNPs from modern human. Sample 140 is a dog. Figure 4 shows, once again, that the Ketchum et al. conclusion above is false. Dog hits are concentrated above 99%ID, human and other primates have much fewer hits above 99%ID. Human results over a variety of DNA analyses can be interpreted in different ways: (1) The sample may be from a normal modern human, (2) the sample may be contaminated with modern human DNA, (3) the sample may be from a feral human with unusual adaptations which are not inherited rather acquired, or (4) the sample may be from a sasquatch which is very human-like and possibly with few mutations, which are not included in the particular sequence.[4] These alternatives are difficult to distinguish without extensive sequencing. Indeed, (1) and (3) may be impossible to distinguish by sequencing alone. On the other hand, human results over some analyses and unknown results over others, is much more likely to be due to contamination, degradation, or coincidence, e.g. foreign sequences in the STR electropherogram. A human or human-like sample should test uniformly human. The temptation to declare a new species based on ambiguous or conflicting results should be resisted. To date no RH has been proven to exist by DNA sequencing. However, “Absence of evidence is not evidence of absence,” as noted by Sykes et al. (2014) and others. All the methods described above are relevant and discriminating if properly applied. They have the potential, especially in combinations, to recognize a previously unknown RH, or to eliminate a suspect candidate as belonging to a known species. Full acceptance of any new RH species may require an accompanying holotype specimen, however, or at the very least very high quality photographic or video evidence. RECOMMENDATIONS What then can be learned from this initial research, and what advice would help future investigators, especially those with limited experience in the use of DNA methods, to achieve their goals of identification? 1. Use forensic techniques in collecting, transporting, and storing samples. The 2013-14 Spike TV series “10 Million Dollar Bigfoot Bounty,” led by biological anthropologist Prof. Todd Disotell and primatologist Natalia Reagan, educated the viewers on sample collection. Prof. Jeff Meldrum’s publications (2006, 2013) are also helpful. Unnecessary contamination only complicates sample preparation, method selection, interpretation of results, and conclusions, at worst making the latter impossible. The subject is complicated enough already. Sterile gloves, sterile forceps, sterile sample containers, and a face mask are essential in the field. Lab protocol and controls to avoid or recognize in-house contamination are well established and described, e.g. in Ketchum et al. (2013). However, the unknown and uncontrollable source of contamination, which is very often forgotten, is what happened to the sample before it was collected. For all practical purposes any environmental sample collected in the field can be assumed to be contaminated, even if separated from its source for only an hour or two or even if removed directly from its source.[5] 2. Begin a DNA project by using a simple mitochondrial species identification method such as cytochrome b, 12S rRNA, or cytochrome c oxidase I with universal primers. The primers should amplify hundreds of base pairs of the sample for the best results. Consult with wildlife forensic experts in selecting these primers. People who only deal with human DNA may not have enough experience in this. Baring extreme contamination, you should then know the species, family, or order (at worst) of the sample, in many cases the former. Expensive “whole genome” sequencing of non-target animals (for example, Ketchum et al. S26 black bear and S140 dog) may thus be avoided. In spite of your best efforts at decontamination, consider that your results may still be affected by contamination. If in doubt, try another mtDNA method (gene) or use well-selected species specific primers to resolve ambiguity before moving on to whole genome sequencing. 3. Use the de novo method of whole genome sequencing, not the reference sequence method if at all possible. Picking a reference sequence biases the results in favor of genes which are conserved between sample and reference. Unless you have other very convincing evidence for a particular species or genus of interest or you are unable to remove contamination, e.g. in blood or saliva samples, de novo sequencing will serve you best. 4. When using BLAST™ there are some guiding principles to be followed: a. One cannot match what is not in the database. A species with relatively shorter sequence entries will be pushed down the hit list (ordered by score), possibly to the point of not being reported as a result (below the “maximum target sequences” lowest score). b. One cannot find what one does not search for. Too narrow search criteria based on preconceived notions can cause false impressions. c. Shorter sequence ranges, can be significant if they match the database very well (99%+). These may not appear or be obvious in preliminary searches because of relatively lower scores. Sort the BLAST™ results (downloaded as an Excel® file) on the %ID. These entries will move toward the top of the hit list. d. If a species level search yields a relatively sparse hit list, expand the search for the suspected species to the genus and/or family level (step back). Good matches to closely related species at these levels may indicate that the species of interest is relatively underrepresented in the database compared to its kin. Compare the total number of database entries for each group through searches of the database by group names. e. Short but contiguous hits can combine to give matches over significantly long sequence ranges. Sort the BLAST® hits by Qstart, smallest to largest (column G), then by Qend, largest to smallest (Column H) to find these. f. A long hit list that contains relatively unrelated species with similar scores is not necessarily the sign of a previously unknown species. It could signal conserved genes, common gene spacers, or that the species of interest is not well represented in the database, if at all (see d). g. Hits with relatively long sequence lengths and high scores can have unacceptably low %ID. Look at the individual %ID numbers in the downloaded Excel® hit list. h. Nearly everything in this is relative (see in previous numbers). Expand the scope of searches to get the proper perspective on scores, matching sequence length, % identity, mismatches, and gaps, especially as they relate to established phylogeny, i.e. the relative similarity of species. i. Nucleotide, Genomes (chromosome), Genome plus Transcription (human), Reference Genomic Sequence and Transcriptome Shotgun Assembly databases should all be searched. The Genome and Reference Genomic Sequence Databases have more sequence information for each species; however, they have much fewer species than the Nucleotide Database. j. The NCBI databases in Genbank are “moving targets,” as new sequence data are entered continually. For example, polar bear sequences were entered in the Nucleotide Database in June, 2013, well after the Ketchum et al. study was published, and were subsequently moved to the Transcriptome Shotgun Assembly Database. On the NCBI homepage search for a target species to see in which databases its DNA resides. k. Scatter charts of %ID, calculated for each hit and displayed across the entire unknown sequence may reveal subtle overall match differences between candidate groups. (Examples: Figs. 3 and 4). Failure to recognize these principles resulted in the Ketchum et al. misidentification of nDNA samples 26 and 140. 5. Phylotrees should first be constructed from BLAST™ hit lists generated without species restrictions from the Genomic Reference Sequence (refseq_genomic) database. This will place the animal in the overall Tree of Life. A subsequent more focused query of a target genus or family may reveal more detailed phylogeny, if these additional sequences are available. Any unusual trees such as Ketchum et al. (2013) Supplemental Figures 5 and 6 should not be accepted. Something is wrong. Bother to look up common names for each unfamiliar scientific (Latin) name in the phylotree. Fish and chickens do not belong on a mammalian phylotree as in Ketchum et al. (2013, Supplemental Figure 5). Mice are not among the closest relatives of humans as in Ketchum et al. (2013, Supplemental Figures 5, 6). 6. Unless you have correctly identified the species by other means, the sample is degradation and contamination free, and the results are above 95%, SNP analysis results are uninterpretable, because low % matches can be attributed to multiple causes. If the above criteria are met, they may be useful in identifying subtle mutations not seen by other sequencing methods. You will have to know which SNPs to look for and what their significance would be, which will not likely be the case until very near the end of any study, if ever. 7. Although a separate type of analysis, microscopic hair analysis often accompanies DNA analysis of hair and so is worthy of a brief discussion here. Light micrographs can be ambiguous or misleading. Recently, their validity has been questioned in court cases, and they are not recommended as evidence for criminal prosecution unless accompanied by DNA analysis (Committee…, 2009). Consider that human hair even has additional features not present in animal hair, such as evidence of bleaching, dying, teasing, spraying, and cutting. More convincing results might be obtained if statistical analyses of numerous samples of various potential target species were performed. However, “No scientifically accepted statistics exist about the frequency with which particular characteristics of hair are distributed in the population. There appear to be no uniform standards on the number of features on which hairs must agree before an examiner may declare a ‘match’” (Committee, 2009). For example, based on overall diameter, medulla diameter, or their ratio, population distributions for each species could determine, always with a certain probability, not a certainty, whether a particular hair belonged to one species or another. Well known statistical techniques are available for this. However, drawing conclusions from a comparison of only two hairs can be dangerous, because populations overlap, and one or both samples may be on their extremes. Ketchum et al. found their suspect sasquatch hair diameters were 80-110 μm, whereas Bindernagel and Meldrum (2012) and Meldrum (2013, “Hair” Chapter) found an average diameter of 65 μm. Notice that the S26 hair photograph in Ketchum et al. (2013, Figure 5B) has the same overall diameter to medulla diameter ratio as the figure of the same S26 in Cassidy et al. (2013). However, Meldrum (2013, “Hair” Chapter) finds that suspected sasquatch samples have no visible medulla. In summary: “The committee found no scientific support for the use of hair comparisons for individualization in the absence of nuclear DNA” (Committee, 2009). Although the Committee was primarily addressing human hair analysis, their conclusion applies to primate hairs as well. It should be noted, however, that primate hairs can be distinguished from other mammals and that they are not differentiated into guard hair and underfur. Conclusion: Use scaled hair micrographs for documentation but not as evidence for a particular species unless part of a very thorough statistical study of multiple suspect species, and even then only as supplemental evidence. Hopefully, as more DNA analyses are performed on RH candidates these recommendations will be modified and/or additional recommendations will be made by those active in the field. [1] Very unfortunately, Khan and White (2012) mislabeled as “A” the haplogroup of their first sample and the three controls in their “Summary of Results” table, which conflicts with their results. A personal communication with Tyler Huggins, the project sponsor, revealed that the authors meant to use “A” as a place holder to indicate that all four samples had the same haplogroup. Recall, above, that they did not specifically mention a haplogroup. Haplogroup (clade) A is an eastern Asia and First American haplogroup, and definitely not what was found and not what they described above. [2] There was some inconsistency here. Fifteen black bear loci were mentioned in the text but only fourteen appeared in the electropherograms. One electropherogram was duplicated, but electropherograms for the loci MU05 and G1A, mentioned in the text, were missing. Also, the G10P locus electropherogram was shown, but the locus was not mentioned in the text. Most likely 14 or 15 of 16 black bear loci were sequenced. Electropherograms showed more than two alleles in some cases, so a second bear may be involved as a contaminant. Alternatively, Justin Smeja’s dog (which found the sample under two feet of snow) could be the source of extra alleles. (More research needed here). As mentioned below, dog nDNA was found in the Ketchum et al. S26 sample (Table 1, Amel X sequence from SGP website), but not recognized by them. [3] A picture of the S35 toenail can be found on the Arizona Cryptozoological Research Organization website: http://www.azcro.net/. The S35 haplogroup H10e was the only one in the study with no extra mutations. By this measure it is as human as human can be. My educated guess (based on a personal experience) is that it is a normal human toenail, discarded by a hiker to the nearby petroglyphs, probably because his boots were too small on a previous hike. [4] Hypertrichosis (Ambras syndrome) is an interesting, relevant example. This defect causes a person to have long hair on his body, which can be localized or general. One form of the disease is linked to the X-chromosome (q24-q27.1) and follows Mendelian genetics (affected father passes only to daughters, affected mother passes to sons and daughters). Other forms are specific to other genes on other chromosomes or are not inherited rather acquired. A hypertrichotic sasquatch would explain (but, of course, is not proven by) human DNA analyses. [5] A living sloth, for example, has multiple species of algae, arthropods, and fungi in its fur. (Pauli et al., 2014). Fortunately, it’s not a candidate species for sasquatch. ACKNOWLEDGEMENTS Thanks go to the Sasquatch Genome Project for making their sequences available online and to Tyler Huggins and Bart Cutino for making their independent lab reports on S26, Khan and White (2012) and Cassidy (2013), respectively, available online. I am also grateful to Prof. Beth Shapiro and Dr. James Cahill for sharing their black bear sequences from Cahill et al. (2013). BLAST™ online help is also appreciated. LITERATURE CITED Altschul SF et al. (1990) Basic local alignment search tool. Journal of Molecular Biology 215(3):403-410. Behar DM et al. (2007) The Genographic Project Public Participation Mitochondrial DNA Database. PLOS Genetics June; 3(6): e104. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1904368/#pgen-0030104-st002 Bindernagel J and Meldrum J (2012) Misunderstandings arising from treating the sasquatch as a subject of cryptozoology. The Relict Hominoid Inquiry 2:81-102. Cahill JA et al. (2013) Genomic Evidence for Island Population Conversion Resolves Conflicting Theories of Polar Bear Evolution. PLOS Genetics March. http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1003345 Coltman D and Davis C (2005) Molecular cryptozoology meets the Sasquatch. Trends Ecology and Evolution 21:60–61. Committee on Identifying the Needs of the Forensic Sciences Community, National Research Council (2009) Analysis of hair evidence. In: Strengthening Forensic Science in the United States: A Path Forward. Washington D.C.: The National Academies Press. pp. 155-161. https://www.ncjrs.gov/pdffiles1/nij/grants/228091.pdf Cronin MA et al. (2014) Molecular phylogeny and SNP variation of polar bears (Ursus maritimus), brown bears (U. arctos), and black bears (U. americanus) derived from genome sequences. Journal of Heredity 105(3): 312–323. Desai NA and Shankar V (2003) Single-strand-specific nucleases. FEMS Microbiological Review 26(No. 5): 457-91. Di Rienzo A and Wilson AC (1991) Branching pattern in the evolutionary tree for human mitochondrial DNA. Proceedings of the National Academy of Sciences USA 88:1597-1601. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC51071/ Edwards CJ and Barnett R (2015) Himalayan ‘yeti’ DNA: polar bear or DNA degradation? A comment on ‘Genetic analysis of hair samples attributed to yeti’ by Sykes et al. (2014). Proceedings of the Royal Society B 282:20141712. http://dx.doi.org/10.1098/rspb.2014.1712 Hailer F et al. (2012) Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science 336:344–347. Hart HV (2016a) Not finding bigfoot in DNA. Journal of Cryptozoology 4: 39-52. Hart HV (2016b) Purported bigfoot mitochondrial DNA: is it modern human? Journal of Cryptozoology (submitted). Hart HV (2016c) Purported bigfoot mtDNA samples have in common rare mutations found in nonhuman primates. Journal of Cryptozoology (submitted). Ketchum MS et al. (2013) Novel North American hominins: next generation sequencing of three whole genomes and associated studies. DeNovo 1:1. Online only: http://sasquatchgenomeproject.org/sasquatch_genome_project_002.htm Kocher TD et al. (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proceedings of the National Academy of Sciences USA 86: 6196-6200. http://www.pnas.org/content/86/16/6196 Lehtinen DA et al. (2008) The TREX1 Double-stranded DNA Degradation Activity Is Defective in Dominant Mutations Associated with Autoimmune Disease. The Journal of Biological Chemistry 283(No. 46):31649–31656. http://www.jbc.org/content/283/46/31649.full Linacre AMT and Tobe SS (2013) Wildlife DNA Analysis: Applications in Forensic Science. Chichester (UK): Wiley-Blackwell. Madden T (2003) The BLAST sequence analysis tool. The NCBI Handbook, Chapter 16. McEntyre J and Ostell J, Eds. Bethesda, MD: National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/books/NBK21097/ Matthiessen P and Laird T (1995) East of Lo Monthong: In the Land of the Mustang. Boston, MA: Shambhala Publishers. McDonald JH (2011) Red hair color: The myth. https://udel.edu/~mcdonald/mythredhair.html Meldrum J (2013) Sasquatch Field Guide: Identifying, Tracking, and Sighting North America’s Relict Hominoid. Arcata: Paradise Cay Publications. Meldrum J (2006) Sasquatch: Legend Meets Science. New York: Tom A. Doherty Associates. Ch. 15 (Also available in Kindle). Melton T et al. (2005) Forensic Mitochondrial DNA Analysis of 691 Casework Hairs. Journal of Forensic Science 50(1):73-80. Melton T and Holland C (2007) Routine Forensic Use of the Mitochondrial 12S Ribosomal RNA Gene for Species Identification. Journal of Forensic Science 52(6):1305-07. Milinkovitch MC et al. (2004) Molecular phylogenetic analyses indicate extensive morphological convergence between the ‘‘yeti’’ and primates. Molecular Phylogenetics and Evolution 31:1–3. https://www.sciencedirect.com/journal/molecular-phylogenetics-and-evolution/vol/31/issue/1 Pauli JN et al. (2014) A syndrome of mutualism reinforces the lifestyle of a sloth. Proceedings of the Royal Society B 281: 20133006. http://rspb.royalsocietypublishing.org/content/royprsb/281/1778/20133006 Starr B (2011) Hair color. The Tech Museum of Innovation. http://genetics.thetech.org/ask/ask400 Sykes BC et al. (2014). Genetic analysis of hair samples attributed to yeti, bigfoot and other anomalous primates. Proceedings of the Royal Society B 281:20140161. http://rspb.royalsocietypublishing.org/content/281/1789/20140161 University Of Alberta (2005) Possible Sasquatch Hair Turns Out To Be Bison. http://www.sciencedaily.com/releases/2005/08/050810133244.htm Ward EJ et al. (1985) Single-strand-specific degradation of DNA during isolation of rat liver nuclei. Biochemistry 24(21):5803-5809. Table 1. Summary of DNA Results from Ketchum et al. (2013) and Independent Laboratories. Hart-Table 1.xlsx Table 2. Extra mtDNA Mutations Not Indicated by Haplogroup. Hart-Table 2.xlsx Figure 3. Sample 26 hits vs. polar bear, dog, human and other primates. Only hits ≥ 95%ID and ≥ 200 bp are plotted. Duplicates removed. Figure 4. Sample 140 hits vs. polar bear, dog, human, and other primates. Only hits ≥ 95%ID and ≥ 150 bp are plotted except >200 bp for polar bear. Duplicates removed. Dr. Haskell V. Hart holds a PhD in chemistry from Harvard University and has a physical, inorganic, and analytical chemistry research background. He was Associate Professor of Chemistry, University of North Carolina at Wilmington, after which he was Senior Staff Research Chemist and Research Manager at Shell Chemicals. At Shell he both conducted analytical research and managed various analytical departments. His research interests have included analytical applications of x-ray diffraction, electron diffraction (two database patents), and gas chromatography-mass spectrometry. Since his retirement, he has focused on long-range detectors and application of DNA sequencing to species identification, especially relict hominoid candidates. His blog, www.bigfootclaims.blogspot.com, contains over thirty articles on this subject and related issues

 1 point
1 point -
PS: Good luck in Maine. My areas are under about 6 feet of snow right now. I need to develop lower elevation "spots" but haven't gotten it done yet. I'm probably on the bench 'til July .. maybe late June if I'm lucky.1 point
-
Actually, animals are surprisingly different in their behavior near death. I grew up in a bear preserve in SW Oregon. We saw a ton of bears, sometimes many in a day, but though we had some big cats, seeing even one in 5 years was unusual. However, so far as remains, I never found the remains of a bear that didn't have a (illegal) bullet hole in it but we found cat remains fairly often. The reason is that bears, sick or dying, seek shelter and privacy, but cats, sick or dying, seek places where they have good visibility around them. It's all a matter of their individual species' comfort zone. So bigfoot ... well, we don't see them lying dead on the forest floor or in other open places. That leaves two options. They could, like bears, hole up in incredibly nasty brush, etc when they are sick or dying. Or the other possibility ... they bury their dead. This behavior has been reported by a few witnesses who claimed to observe the burials happening. Part of our search, then, also includes finding places where the ground is disturbed over an area large enough to put a sasquatch under. This could be nearly anywhere but I'd expect it to be pretty private else someone would have noticed the ground being disturbed and dug one up by now. So far, just guesses though, and no success at the end of any of them yet. It should be enough to make a person open-minded about some of the woo rabbit holes. Not necessarily go down them, but at least walk around the hole and give it a good hard look. Because, like I said, at least some of our assumptions have to be wrong else we would not still have an unsolved puzzle. MIB1 point
-
Sure, something isn't right. That said, I do not think there is any broad conspiracy to conceal. I suspect at least one, probably several, grossly flawed assumptions that lead us to look somewhere besides where the answers lie. One possibility is that there are 2 or more sources to the reports rather than just one. So long as we insist on blindly looking for that one we perceive, we are looking in the gap where there is nothing between the two actual sources. I think we need to review our data, return to the observations, discard the interpretations, and start over from scratch. Instead of filtering evidence to support the answer we want to find, I think we should look at all of the evidence to see what it actually shows us.1 point
-
Yeah I will. I also caught your youtube channel and started watching there too. Very entertaining. Just the fact that you are investigators and investigating and reporting on it in near real time is cool. I didn't realize you had just released that Fallen Worlds chapter 1 video a week or so before I posted the younger dryas post in the general forum. It's an interesting story.1 point
-
Bigfoot is a mammal, females have breasts, they certainly breast feed.1 point
-
Thank you for posting this article. I know that when I print it out, and then read it, much more will sink in. It's just the way my brain works!1 point
-
I've listened to a few of these podcasts and am really starting to get addicted. I'll be binging on them while working.1 point
-
I had a high-school girlfriend, a cheerleader. Good looking girl, well endowed, during games. Oddly, became a little less "well-figured" after changing from her cheerleading sweater. As if there weren't already enough life mysteries to a young Incorrigible.1 point
-
Hmmm... unless it's like eyespot mimicry in certain species to deceive predators. Boob mimicry.1 point
-
Some claim to. Most of them don't believe in them. Scoftics /skeptics .1 point
-
😉 Bigfootery Today is a mythological magazine promoting the research efforts of Bigfooters everywhere.1 point
-
If any one is interested in the the episode we recorded for the podcast heres a link: https://www.wildandweirdwv.com/podcast/episode/4c328091/bigfoot-investigation-wineberry-site1 point









This leaderboard is set to New York/GMT-04:00